Capítulo 19.
Genética humana: Pasado, Presente y Futuro
Los principios de la genética son, por
supuesto, los mismos para los seres humanos que para los miembros de cualquier
otra especie eucariótica diploide. En la práctica, sin embargo,
hay algunas diferencias importantes. El número diploide normal de cromosomas
en la especie humana es 46: 44 autosomas y 2 cromosomas sexuales, XX en las
mujeres y XY en los varones.
Es posible determinar
el número, el tamaño y la forma de los cromosomas de una célula
somática de un organismo determinado. El ordenamiento sistematizado de
los cromosomas de un organismo determinado constituye su cariotipo .
Numerosos estudios sobre
la determinación del sexo llevó a concluir que en muchas especies
esa determinación tiene una base genética. Este concepto, a su
vez, llevó a identificar un gen de la iniciación de la determinación
del sexo en los machos. Esta función ha sido asignada al gen SRY (Región
determinante del Sexo del cromosoma Y) localizado en el cromosoma Y de los seres
humanos. También se postula la existencia de otros genes implicados en
la determinación del sexo.
En el momento del nacimiento
de un individuo se presentan alteraciones estructurales o funcionales que pueden
manifestarse tanto en distintas etapas de la vida y se denominan defectos congénitos.
Estos defectos pueden tener una base genética o no. Los que no son hereditarios
pueden ser consecuencia de la acción de factores ambientales que alteren
el desarrollo embrionario.
La tecnología del
DNA recombinante está aportando nuevos medios para el diagnóstico
precoz de las enfermedades hereditarias. Entre las herramientas más importantes
para este diagnóstico están los RFLPs (polimorfismos de longitud
de fragmentos de restricción) y las sondas radiactivas.
El DNA también
se está usando en forma terapéutica, en la denominada terapia
génica. La administración del DNA como medicamento puede, al menos
teóricamente, corregir enfermedades genéticas, enlentecer la progresión
de tumores, enfrentar infecciones virales y detener enfermedades neurodegenerativas.
Es decir, puede dirigirse tanto a enfermedades hereditarias como a afecciones
adquiridas.
Muchos laboratorios de
todo el mundo están sumando sus esfuerzos para mapear y secuenciar el
genoma humano completo. Se espera que este proyecto pueda responder muchas preguntas
desconcertantes. Si la experiencia pasada sirve de guía, también
es probable que haga surgir muchas preguntas nuevas.
El cariotipo humano
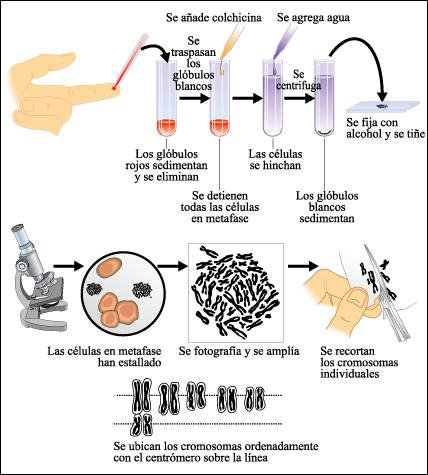
La clasificación de los cromosomas para identificar las anormalidades morfológicas y numéricas más importantes se lleva a cabo en un número creciente de centros genéticos médicos. El resultado del procedimiento es una demostración gráfica del complemento cromosómico -o dotación cromosómica- conocida como cariotipo. Los cromosomas que se muestran en un cariotipo son cromosomas en metafase de la mitosis, cada uno de los cuales consiste en dos cromátides hermanas unidas por sus centrómeros. Para preparar un cariotipo, el proceso de división celular se interrumpe en la metafase, añadiendo colchicina, una droga que evita los siguientes pasos de la mitosis, ya que interfiere con los microtúbulos del huso. Después del tratamiento y de la tinción, los cromosomas se fotografían, se amplía la fotografía, se los recorta y se los ordena de acuerdo con su tamaño. Los cromosomas del mismo tamaño se aparean según la posición del centrómero, lo que evidencia la presencia de "brazos" de distinta longitud. A partir del cariotipo pueden detectarse ciertas anormalidades, como la aparición de un cromosoma o de un segmento cromosómico supernumerario.
Preparación de un cariotipo humano
Si bien se puede determinar el número, el tamaño y la forma de los cromosomas e identificar los pares homólogos presentes en la metafase mitótica de una célula somática de un organismo determinado, algunos de los cromosomas más pequeños son bastante semejantes en su morfología. En estos casos, la tinción para mostrar patrones de bandas posibilita distinguir cromosomas del mismo tamaño e identificar los homólogos.
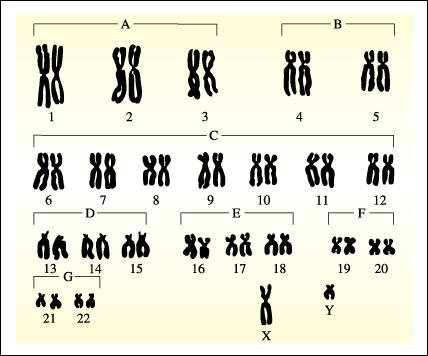
Cromosomas de una sola célula somática ordenados formando un cariotipo.
Los patrones de bandas que caracterizan los cromosomas no son visibles en este esquema. En un cariotipo, los autosomas se agrupan en tamaños (A, B, C, etc.) y se les adjudican los homólogos probables. El número diploide normal de cromosomas de la especie humana es de 46: 22 pares son autosomas y 2 son cromosomas sexuales. Una mujer normal tiene dos cromosomas X y un hombre normal tiene un X y un Y, como se muestra aquí.
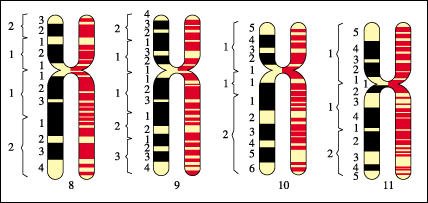
Mapa estándar de patrones de bandeo de los cromosomas 8 a 11 en el cariotipo humano, determinado en la etapa metafásica (bandas negras) y en la profase temprana de la mitosis (bandas coloreadas).
Los cromosomas en la profase temprana son mucho más largos y delgados que los cromosomas en la metafase y, por lo tanto, pueden detectarse muchas más bandas. Todas las bandas que se muestran aquí se tiñen con un reactivo específico. Nótese cómo estos esquemas de cromosomas, que son semejantes en tamaño y forma, pueden ser distinguidos rápidamente por sus patrones de bandeo.

Cromosomas de una célula somática en división de una mujer, fotografiados después de la replicación y condensación.
Los cromosomas que se ven en la figura han sido teñidos y las distintas
bandas que se observan facilitan la identificación de los pares homólogos.
La estructura redondeada y amarilla que se muestra en la parte superior derecha
del esquema es un núcleo interfásico.
Defectos congénitos
Los defectos congénitos
son alteraciones estructurales o funcionales que están presentes en el
momento del nacimiento y pueden manifestarse tanto en los primeros días
de vida como en etapas posteriores. Estos defectos pueden tener una base genética
o no. Los que no son hereditarios, pueden ser consecuencia de la acción
de factores ambientales que hayan alterado el desarrollo embrionario. Entre
estos factores se encuentra la ingestión excesiva por parte de la madre
de algunos medicamentos -como ciertos tranquilizantes y algunos antibióticos-
o de alcohol. La exposición a agentes físicos, como las radiaciones,
o la acción de agentes infecciosos, también puede alterar el desarrollo
del feto.
Entre las enfermedades que tienen
base genética, se encuentran las que son producidas por alteraciones
en los cromosomas, las debidas a la alteración de un gen principal y
las causadas por alteraciones multifactoriales que incluyen factores genéticos
y ambientales.
Las anormalidades cromosómicas
visibles incluyen alteraciones numéricas, habitualmente resultado de
la no disyunción, y alteraciones estructurales como las translocaciones
y las deleciones. El síndrome de Down está entre los trastornos
asociados con alteraciones numéricas; puede ser causado o bien por no
disyunción o, menos comúnmente, por una translocación.
Los cromosomas sexuales supernumerarios pueden ser resultado de la no disyunción
y suelen estar asociados con la esterilidad.
Muchas enfermedades genéticas
son el resultado de deficiencias o defectos en las enzimas u otras proteínas
críticas. Éstos, a su vez, están causados por mutaciones
en los genes que codifican las proteínas. Cuando los alelos que resultan
de las mutaciones son recesivos, las enfermedades aparentes sólo afectan
a los individuos homocigotas. Estas enfermedades incluyen fenilcetonuria, enfermedad
de Tay-Sachs, anemia falciforme y algunas otras asociadas con variaciones en
las moléculas de la hemoglobina. Las enfermedades genéticas causadas
por alelos autosómicos dominantes incluyen una forma de enanismo y la
enfermedad de Huntington.
Los defectos genéticos
se expresan con mucha mayor frecuencia en varones que en mujeres y son causados
por alelos mutantes en el cromosoma X. Los alelos mutantes responsables de las
características ligadas al sexo, habitualmente son recesivos respecto
de los alelos normales y no se expresan en las mujeres heterocigotas quienes,
sin embargo, pueden transmitirlos a sus hijos. Las características ligadas
al sexo incluyen: ceguera a los colores, hemofilia y distrofia muscular de Duchenne.
Las enfermedades multifactoriales
están ocasionadas por una predisposición genética determinada
por la combinación de varios genes que interactúan con factores
medioambientales diversos. El diagnóstico de las enfermedades genéticas
comienza con un estudio del cuadro clínico del paciente que incluye sus
antecedentes familiares. En la actualidad, es posible el diagnóstico
prenatal de diversas enfermedades por medio de diferentes métodos. Algunos
se denominan no invasivos y comprenden: la ecografía clásica,
la identificación de ciertas sustancias en el suero materno y la ecografía
de alta resolución. Otros, llamados invasivos, incluyen la amniocentesis
y la biopsia de vellosidades coriónicas. Estos últimos métodos
permiten llevar a cabo el diagnóstico prenatal citogenético.
Diagnóstico de las enfermedades
genéticas
El diagnóstico de las
enfermedades genéticas comienza con un estudio del cuadro clínico
del paciente, que incluye sus antecedentes familiares.
Algunas enfermedades genéticas
pueden ser detectadas en etapas tempranas, lo que incrementa las posibilidades
de su prevención o tratamiento. En la actualidad, es posible el diagnóstico
prenatal de ciertas enfermedades por medio de diferentes métodos.
Algunos de ellos, como la ecografía,
la identificación de ciertas sustancias (por lo general proteínas)
en el suero materno y la ecografía de alta resolución son no invasivos.
Otros métodos son invasivos
e incluyen la amniocentesis y la biopsia de vellosidades coriónicas que
permite llevar a cabo el diagnóstico prenatal citogenético.
Sin embargo, en la mayoría de los casos, el tratamiento de las enfermedades detectadas por pruebas prenatales todavía no es posible, y los padres se enfrentan con la difícil decisión de abortar o no el feto afectado.
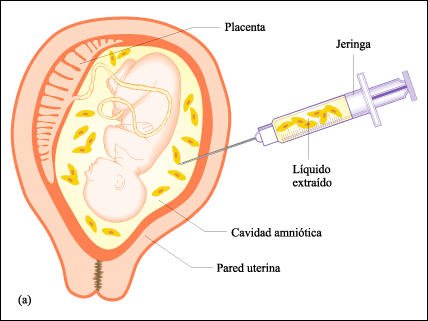
Amniocentesis.
En la amniocentesis, primero se determina la posición del feto con ultrasonido,
con la ecografía permanentemente proyectada en una pantalla de televisión.
Entonces se efectúa la punción y se extrae de la cavidad amniótica
líquido con células fetales. Las células se cultivan y
se estudian para encontrar alteraciones cromosómicas y otros desórdenes
genéticos. La prueba no se realiza hasta la semana 16 de gestación
para asegurar que habrá células fetales suficientes y para que
la cantidad de líquido extraído -aunque es poca- no perjudique
al feto.
En la ecografía del útero de una mujer gestante con un feto de
cuatro meses. Las manchas negras dentro de la pared muscular del útero
son el líquido amniótico que envuelve al feto. El feto yace sobre
su espalda, con la cabeza situada a la izquierda y parece que se chupa el pulgar.
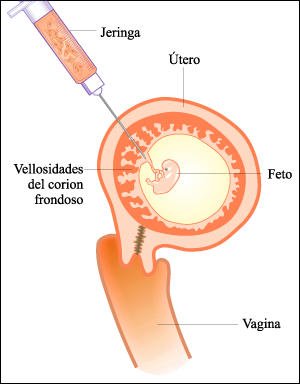
Biopsia de vellosidades coriónicas.
En la biopsia de vellosidades
coriónicas, primero se toman las muestras con una jeringa que se introduce
a través de la pared abdominal. El proceso se monitorea por medio de
una ecografía, de la misma forma que en la amniocentesis.
Desde el punto de vista de la
genética humana, una de las principales recompensas de la tecnología
del DNA recombinante ha sido la capacidad de diagnosticar muchas de las enfermedades
hereditarias mencionadas.
Existen marcadores que se conocen
como RFLPs (se pronuncia "rif-lips" y se traduce como "polimorfismos
de longitud de los fragmentos de restricción").
Los RFLPs son el resultado de variaciones naturales -mutaciones- que eliminan o alteran la secuencia de reconocimiento para una enzima de restricción. Cuando esta mutación está asociada con un alelo que causa una enfermedad genética, puede suministrar un marcador diagnóstico para ese alelo.
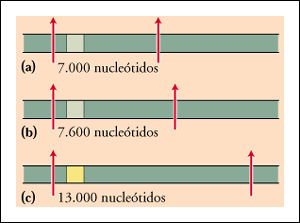
Una prueba, usando RFLPs para detectar la presencia del alelo de la anemia falciforme.
Al tratar el DNA humano con la enzima de restricción HpaI se producen
tres fragmentos de restricción posibles que contienen el gen para la
cadena beta de la hemoglobina. En personas con el alelo normal (gris) para esta
proteína, los fragmentos tienen 7.000 o 7.600 nucleótidos de largo.
En los individuos que llevan el alelo para la anemia falciforme, falta una secuencia
de reconocimiento para la enzima de restricción HpaI (presente en el
DNA de las personas con el alelo de la cadena beta de la hemoglobina normal).
Como consecuencia, el fragmento generado es mayor (13.000 nucleótidos).
El ligamiento entre el alelo para anemia falciforme y la pérdida del
sitio para HpaI vale sólo para las poblaciones de África Occidental
o que fueron originarias de esta zona. En cambio, entre los individuos de África
Oriental o provenientes de ella, el alelo de la anemia falciforme está
asociado con el fragmento de 7.600 nucleótidos.
Las sondas radiactivas constituyen
otra herramienta para llevar a cabo la localización y aislamiento de
ácidos nucleicos. Estas sondas son fragmentos cortos de DNA o de RNA
de cadena simple, marcados con un isótopo radiactivo que se unen al alelo
normal o mutante y pueden ser usadas para la detección y el diagnóstico.
En este caso, para sintetizar cada sonda es necesario conocer la secuencia del
alelo.
Tratamiento de las enfermedades
genéticas
A lo largo de los últimos
siglos la medicina ha experimentado cambios revolucionarios. Los nuevos conocimientos
y prácticas surgidos de la introducción de la microscopia, la
anestesia, la vacunación, los antibióticos y los trasplantes son
testimonio de estas transformaciones. La medicina se prepara ahora para un giro
sustancial: la utilización terapéutica del DNA, denominada terapia
génica. La administración del DNA como medicamento puede, al menos
teóricamente, corregir enfermedades genéticas, enlentecer la progresión
de tumores, enfrentar infecciones virales y detener enfermedades neurodegenerativas.
Es decir, puede dirigirse tanto a enfermedades hereditarias como a afecciones
adquiridas.
La mayor parte de los ensayos
de terapia génica propuestos sólo intentan suplementar con el
gen útil un tipo seleccionado de células, para compensar la falta
o el defecto en ese gen o aportar una nueva característica. Muchas terapias
contra el cáncer intentan inducir a las células cancerosas a producir
sustancias que las eliminen directamente, despierten una fuerte respuesta inmune
contra ellas o anulen el aporte sanguíneo que los tumores necesitan para
crecer. Los problemas técnicos que enfrenta la aplicación de la
terapia génica son importantes e impiden que los resultados sean los
deseados. Uno de los problemas es la baja eficiencia de los métodos de
administración del DNA a las células blanco. Los genes se proveen
básicamente de dos maneras; en ambos casos, los genes son primero puestos
en "transportadores" o vectores -por ejemplo, virus- capaces de depositar
el gen dentro de las células. En el método más común,
los científicos remueven células de un tejido seleccionado del
paciente, las exponen al vector que porta el gen en el laboratorio (ex vivo)
y luego retornan las células corregidas genéticamente al individuo.
El otro método consiste en introducir el vector portador del gen corrector
directamente en el cuerpo (in vivo), por lo general en el tejido por tratar.
En el cuerpo, ciertos genes son útiles sólo si su expresión
es regulada de tal manera que se sintetice la cantidad apropiada de proteína
en el momento adecuado. Para muchas aplicaciones de la terapia génica,
esa regulación tan precisa no es necesaria. Por ejemplo, para corregir
trastornos de la coagulación, como la hemofilia, todo lo que se necesita
es lograr un nivel adecuado de la proteína que interviene en este proceso.
Esta proteína puede ser provista tanto por las células hepáticas
como por otras células (células musculares, fibroblastos o células
de la sangre).
El desarrollo de estas estrategias ha suscitado muchas controversias, no sólo entre los científicos, sino también en diferentes sectores de la sociedad. Muchas opiniones advierten sobre los posibles riesgos de introducir DNA foráneo en el genoma de un individuo, ya que sabemos que los genes no están aislados, sino que interactúan entre sí. En la medida en que se avance tanto en la investigación clínica como en la investigación básica, probablemente se irán despejando algunas de esas dudas.
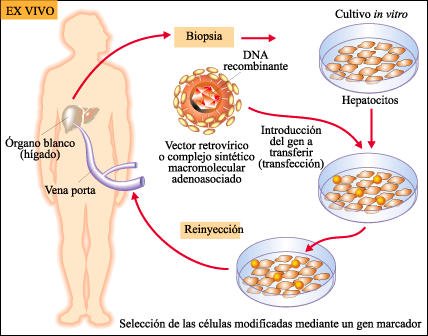
En la terapia génica se utilizan dos estrategias principales.
Una de las estrategias de la terapia génica consiste en extraer células de un paciente, cultivarlas y modificarlas in vitro -generalmente utilizando un vector viral-.Luego se reimplantan esas células en el paciente. En este caso, el riesgo de rechazo del implante por parte del sistema inmunitario es mínimo. Esta estrategia se denomina ex vivo y es la más practicada hasta el momento.
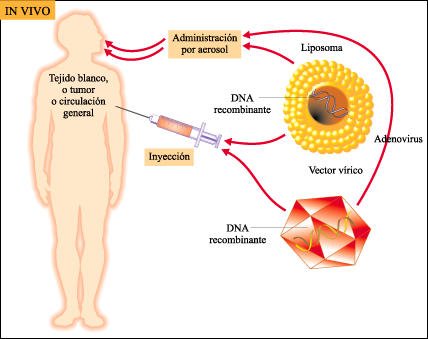
Otra estrategia de terapia génica.
Una segunda estrategia de terapia génica consiste en administrar el gen
"corrector" al paciente in vivo. Con esta estrategia, sin embargo,
no es posible controlar la eficacia de la transferencia del gen.
El proyecto genoma humano: genética
y sociedad
En la actualidad se está
llevando a cabo la mayor y más ambiciosa de las empresas acometidas alguna
vez en investigación médica o biológica: mapear y secuenciar
todo el genoma humano.
El objetivo más importante
del denominado Proyecto Genoma Humano es determinar la secuencia completa de
nucleótidos del DNA humano e identificar y localizar los genes en los
cromosomas .
Para lograr esta empresa, se
mapean los genes a través de marcadores y se los localiza en los cromosomas
por medio de diferentes técnicas moleculares. Por otra parte, se determina
la secuencia de esos genes. En los últimos años, las técnicas
de secuenciación recibieron un impulso importante con el advenimiento
de métodos automáticos, cada vez más eficientes.
Si bien el genoma humano presenta
polimorfismos -lo que hace que la secuencia de DNA de cada persona sea diferente-,
hay secuencias generales que se reconocen en todos los individuos. Una vez que
un gen ha sido secuenciado en varias personas, se pueden identificar las secuencias
polimórficas e intentar relacionarlas con la susceptibilidad a una determinada
enfermedad. Por otra parte, la identificación de secuencias homólogas
y la comparación con las de organismos modelo ampliamente estudiados,
permite inferir las posibles funciones de un gen.
El 26 de junio de 2000 se presentó
en sociedad el primer "borrador" con la secuencia completa del genoma
humano. Esta fecha será recordada en la historia de la biología.
Sin embargo, no es suficiente
conocer la descripción completa del genoma humano para comprender y curar
las enfermedades, ya que la secuencia de nucleótidos no alcanza para
comprender la organización funcional de los genes. Por lo tanto, nuevas
tecnologías y herramientas conceptuales deben encausarse de tal modo
que promuevan el estudio sistemático de la función de cada gen:
una transición del estudio estructural del genoma al estudio funcional.
También, a medida que se acerca el momento de finalización del
proyecto, hay una creciente excitación acerca de qué podría
revelar la lectura de este "libro del hombre", como ha sido llamado.
El Proyecto Genoma Humano lleva
al planteo de ciertas consideraciones éticas. Muchas personas, desde
distintos sectores sociales, han alertado acerca del riesgo potencial que lleva
implícito este proyecto. Es posible que los resultados de esta enorme
empresa, lejos de utilizarse para mejorar las condiciones de vida humana, puedan
llegar a emplearse con otros fines y resultar perjudiciales para muchas personas.


